Актуальные вопросы диагностики муковисцидоза
Статьи
![]()
ЖУРНАЛ «ПРАКТИКА ПЕДИАТРА»
Опубликовано в журнале:
«ПРАКТИКА ПЕДИАТРА»; март-аперль; 2015; стр. 20-27.
Е.И. Кондратьева, д. м. н., профессор, В.Д. Шерман, к. м. н., Н.И. Капранов, д. м. н., профессор, Н.Ю. Каширская, д. м. н., профессор, НКО муковисцидоза ФГБНУ «МГНЦ», ГБУЗ «ДГКБ № 13 им. Н.Ф. Филатова ДЗМ», г. Москва
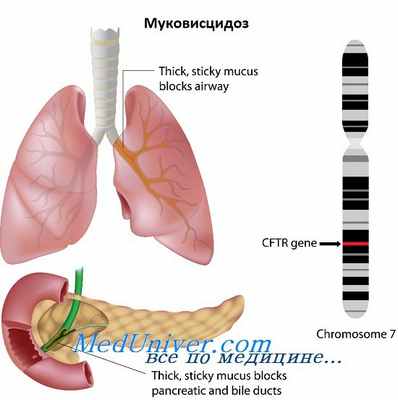
Муковисцидоз (МВ), или кистозный фиброз (cysticfibrosis), — одно из наиболее частых моногенных наследственных заболеваний с полиорганной патологией, резко сокращающее продолжительность и качество жизни пациентов без адекватного комплексного лечения в течение всей жизни. МВ распространен среди населения всей Земли, но наиболее часто поражает европеоидов: в среднем с частотой 1 на 2500-4500 новорожденных. Еще совсем недавно больные муковисцидозом умирали в раннем детском возрасте или даже на первом году жизни от пневмонии и истощения, обусловленными мальабсорбцией.
Ключевые слова: диагностика, генетика, мутации, неонатальный скрининг, потовая проба, эластаза кала.
Key words: cystic fibrosis, diagnosis, genetics, mutation, newborn screening, sweat test, fecal elastase.
Болезнь прежде всего характеризуется повышенной продукцией вязкого бронхиального секрета, частыми легочными инфекциями и обструкцией дыхательных путей. По мере прогрессирования легочной болезни образуются участки ателектазов, развивается эмфизема, постепенно разрушается паренхима легких с развитием бронхоэктазов и участков пневмосклероза, а больной имеет высокий риск погибнуть от легочно-сердечной недостаточности. В финальной стадии заболевания пересадка комплекса «сердце-легкие» остается для больного единственной надеждой. Помимо бронхолегочной системы у большинства больных муковисцидозом поражается поджелудочная железа, при этом это происходит внутриутробно. Недостаточность панкреатических ферментов обусловливает нарушение всасывания жиров и белков, развитие нутритивной недостаточности. В результате больные отстают в росте и страдают гипотрофией. Продукция инсулина также может быть нарушена, что ведет к развитию диабета. К частым осложнениям течения муковисцидоза относят остеопороз, а также жировой гепатоз с переходом в цирроз. При наличии «мягкой» мутации клинические проявления развиваются постепенно, преобладают моносимптомы, диагноз «муковисцидоз» устанавливается поздно или случайно.
Своевременная диагностика муковисцидоза, обеспечивающая в большинстве случаев раннее начало терапии, в том числе на доклиническом этапе, улучшает прогноз заболевания, повышает эффективность лечения, позволяет предупредить развитие тяжелых осложнений, значительного отставания в физическом развитии, а в ряде случаев и необратимых изменений в легких. Ранняя диагностика позволяет семье вовремя решить необходимые вопросы, связанные с рождением здорового ребенка (генетическое консультирование, пренатальная диагностика МВ в последующие беременности).
Диагностика делится на:
1) пренатальную диагностику;
2) диагностику по неонатальному скринингу (до клинических проявлений или при их дебюте);
3) диагностику при клинических проявлениях:
4) диагностику среди родственников больных.
В настоящее время налаживается дородовая диагностика муковисцидоза в перспективных и информативных семьях (Москва, Санкт-Петербург, Уфа, Томск, Красноярск, Ростов-на-Дону, Владивосток и некоторые другие города), что, безусловно, важно для профилактики этой тяжелой патологии. Пренатальная диагностика возможна в виде ДНК-диагностики при проведении амниоцентеза (получение околоплодных вод в ранний срок -13-14 недель и поздний — обычно 16-20 недель беременности) в семье носителей одной мутации гена CFTR и имеющей больного ребенка. Диагноз может быть заподозрен при УЗИ плода внутриутробно при наличии характерной УЗ-характеристики в виде гиперэхогенного кишечника. УЗИ во время беременности рекомендуют в скрининговые сроки: 11-14, 18-21 и 30-34 недели беременности. Обязательно проводят повторное исследование. В 50-78% случаев это состояние будет связано с МВ и проявится мекониальным илеусом. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высокоспецифичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями. При этом ДНК-диагностика родителей дает необходимую информацию о наличии мутаций у каждого из родителей и позволяет предполагать заболевание у ребенка при рождении.
Клинические признаки
1. Диагностика классической формы МВ обычно не представляет сложностей. Классический фенотип больного является результатом наличия двух мутантных копий гена муковисцидозного трансмембранного регулятора (CFTR) и характеризуется хронической бактериальной инфекцией дыхательных путей и придаточных пазух носа, стеатореей из-за внешнесекреторной недостаточности поджелудочной железы, мужским бесплодием из-за обструктивной азооспермии, а также повышенной концентрацией хлоридов потовой жидкости.
2. Проблемы диагностики МВ, как правило, связаны с фенотипическим разнообразием его форм, обусловленным генетическим полимор-
В ряде случаев атипичного течения МВ возможна его диагностика во взрослом возрасте. Как правило, в этой группе больных отмечается более мягкое течение болезни в связи с сохранностью функции поджелудочной железы и нетяжелым поражением органов дыхания.
В абсолютном большинстве случаев МВ может быть диагностирован в раннем детском возрасте (в 90% случаев — на первом году жизни). К сожалению, нередки случаи диагностики МВ у взрослых с классическим фенотипом.
Диагностика МВ у носителей «мягких» генотипов (актуально для детей, рожденных до 2006-2007 гг., и взрослых):
В настоящее время выделяют несколько групп риска по МВ.
Основной группой риска по заболеванию в РФ в настоящее время являются новорожденные с неонатальной гипертрипсиногенемией. Учитывая возможность получения ложноотрицательных результатов неонатального скрининга, а также то обстоятельство, что в РФ неонатальный скрининг на МВ проводится с 2006-2007 гг., не теряет своей актуальности анализ групп риска, включающих пациентов с патологией желудочно-кишечного тракта, бронхолегочными нарушениями, патологией других органов и родственников больных МВ (табл. 1).
Таблица 1.
Группы риска для дифференциальной диагностики муковисцидоза
| I. Бронхолегочные нарушения |
| 1. Повторные и рецидивирующие пневмонии с затяжным течением, особенно двусторонние 2. Бронхиальная астма, рефрактерная к традиционной терапии 3. Рецидивирующие бронхиты, бронхиолиты, особенно с высевом Ps. aeruginosa 4. Двусторонние бронхоэктазы |
| II. Изменения со стороны желудочно-кишечного тракта |
| 1. Синдром нарушенного кишечного всасывания неясного генеза 2. Мекониальный илеус и его эквиваленты 3. Гиперэхогенность кишечника плода 4. Желтуха обструктивного типа у новорожденных с затяжным течением 5. Цирроз печени 6. Сахарный диабет 7. Гастроэзофагеальный рефлюкс 8. Выпадение прямой кишки |
| III. Патология со стороны других органов |
| 1. Нарушение роста и развития 2. Задержка полового развития 3. Мужское бесплодие 4. Хронический синусит 5. Полипы носа 6. Электролитные нарушения |
| IV. Члены семей больных муковисцидозом |
Среди клинических проявлений, характерных для МВ, можно выделить высоко-и менее специфичные (табл. 2). Состояния, представленные в левой колонке таблицы, в абсолютном большинстве случаев встречаются у больных МВ. Причиной состояний из правой колонки могут быть другие заболевания, например первичная цилиарная дискинезия, гуморальный иммунодефицит и т. д.
Таблица 2.
Клинические проявления, характерные для МВ
| Высокоспецифичные для МВ | Менее специфичные для МВ |
| Желудочно-кишечные:
|
Желудочно-кишечные:
|
| Со стороны дыхательных путей:
|
Со стороны дыхательных путей:
|
| Другое:
|
Другое:
|
В таблице 3 представлены особенности проявлений МВ в разные возрастные периоды. Знание этих особенностей помогает специалистам, наблюдающим пациента с теми или иными симптомами, включить МВ в перечень заболеваний для дифференциальной диагностики. Особенно это касается детей раннего возраста, когда клиническая картина еще может быть неполной, но на себя будут обращать внимание некоторые проявления, например мекониальный илеус при рождении или синдром потери солей, не имеющий связи с патологией почек. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высоко специфичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями.
Таблица 3.
Клинические особенности проявлений МВ в различные возрастные периоды
| 0-2 года | |
|
|
|
| 3-16 лет | |
|
|
|
Диагностические критерии МВ
Для решения проблем диагностики МВ, в том числе и его атипичных форм, были разработаны критерии, согласно которым обязательным для МВ является наличие характерного клинического синдрома плюс доказательство какого-либо нарушения функции хлорного канала.
Учитывая все научные достижения в понимании природы муковисцидоза и МВ-зависимых заболеваний за последние 10 лет, в 2013 году группа экспертов Европейского общества муковисцидоза (European Cystic Fibrosis Society) под руководством Carlo Castellani подготовила новые стандарты диагностики в редакции Alan R. Smyth и Scott Bell (схема).
Схема.
Диагностические критерии муковисцидоза ECFS 2013
| Положительная потовая проба и/или две мутации МВТР, вызывающие МВ (согласно базе CFTR-2) |
И | Неонатальная гипертрипсиногенемия или характерные клинические проявления, такие как диффузные бронхоэктазы, высев из мокроты значимой для МВ патогенной микрофлоры (особенно синегнойной палочки), экзокринная панкреатическая недостаточность, синдром потери солей, обструктивная азооспермия |
Неонатальный скрининг
Проводится на основании Методических рекомендаций по проведению неонатального скрининга в РФ с использованием Европейских рекомендаций по неонатальному скринингу. 90% новорожденных без клинических проявлений муковисцидоза диагноз может быть установлен на основании скрининга в возрасте до 6 недель. В 5-10% случаев возникают трудности с диагностикой муковисцидоза (Cystic Fibrosis Foundation Patient Registry, 2005 Annual Data Report to the Center Directors. Bethesda, MD: CFF).
Проблемы неонатального скрининга:
Потовая проба
Показания:
1. При положительном результате неонатального скрининга (двукратном повышении уровня иммунореактивного трипсиногена в крови в течение первого месяца жизни ребенка).
2. При наличии у пациента каких-либо характерных клинических проявлений МВ.
3. Случаи МВ в семье.
Потовая проба является надежным методом диагностики МВ у 98% больных. Исследование можно проводить всем детям через 48 часов после рождения, хотя у новорожденных могут быть проблемы с набором пота. Несмотря на то, что «золотым стандартом» диагностики МВ считается количественное определение хлоридов в потовой жидкости (классический метод Гибсона — Кука), метод определения проводимости на аппаратах «Макродакт» и «Нанодакт» («Вескор», США) показал хорошую с ним корреляцию в многочисленных исследованиях.
Оценка результата
При положительном результате потовой пробы (хлориды > 60 ммоль/л при классическом методе Гибсона — Кука и/или проводимость > 80 ммоль/л NaCl) диагноз подтверждается.
Генетическое исследование
Генетическое исследование проводится после потовой пробы. Однако в связи с ограниченными возможностями ДНК-диагностики в России данный метод не является обязательным, однако применяется с исследовательской целью и для окончательного подтверждения диагноза.
На первом этапе ДНК-обследования наиболее часто используется панель, включающая 28 мутаций, как наиболее частых в мире, так и специфичных для России: F508del, CFTRdele2,3(21kb), 3849+10kbC>T, W1282X, 2143delT, 2184insA, 1677delTA, N1303K, G542X, R334W, E92K, L138ins, 394delTT, 3821delT, S1196X, 2789+5G>A, G85E, 2183AA>G, 604insA, 621+1G>T, R117H, R347P, R553X, 3667insTCAA, G551D, I507del, 1717-1G>A, 2184delA. По данным лаборатории генетической эпидемиологии ФГБУ «Медико-генетический научный центр» (МГНЦ) РАМН, при использовании данной панели удается обнаружить лишь около 82,5% мутантных аллелей у больных МВ. В случае когда при положительной потовой пробе не будет найдено ни одной мутации гена (что само по себе маловероятно), может потребоваться секвенирование гена МВ, позволяющее идентифицировать примерно 98% мутаций в гене CFTR.
Рекомендации:
1. На основании данных национального регистра больных МВ по ДНК-диагностике гена CFTR установлены особенности характера и частоты мутаций в регионах страны. На основе данных регистра рекомендуется создание региональных рекомендаций по определению мутаций со ссылкой на регистр (последнюю версию).
2. Отсутствие мутациий без проведения секвенирования — недостаточно для исключения МВ.
3. Некоторые мутации МВТР (3849+10 kb C>T) ассоциированы с нормальным или пограничным результатом потового теста.
4. «Мягкие» мутации характеризуются поздним дебютом заболевания, пограничным значением потовых проб, выявляются чаще при секвенировании.
5. Пациенты с пограничными результатами потовых проб (хлориды 30-60 ммоль/л и/или проводимость 50-80 ммоль/л), единственной мутацией гена представляют реальные трудности для диагностики.
Для диагностики МВ или его исключения при пограничных результатах пробы необходимо:
В европейских странах для подтверждения дефекта ионного транспорта применяется метод определения разности назальных потенциалов или измерение электрического тока в биоптате кишки, отражающие нарушение функции хлорного канала. Оба метода основаны на электрическом характере транспорта ионов и являются высокоинформативными для диагностики МВ.
Диагностика панкреатической недостаточности включает:
У больных МВ показатель эластазы может снижаться в течение первых лет жизни, поэтому определяется в динамике. Низкий уровень панкреатической эластазы расценивается как один из признаков МВ. Приблизительно 1% пациентов с МВ имеет пограничный результат потового теста в комплексе с сохранной функцией поджелудочной железы и хроническим бронхитом.
Диагностика хронического бронхолегочного процесса:
В качестве дополнительных диагностических маркеров могут быть использованы азооспермия в постпубертатном возрасте, идентификация МВ-ассоциированных патогенов из респираторного тракта, рентгенологические признаки синусита.
Знание основных симптомов МВ и особенностей его течения в разные возрастные периоды позволяет своевременно заподозрить наличие заболевания и направить пациента для дальнейшего обследования. Нередкие случаи поздней диагностики МВ связаны как с отсутствием у врачей достаточных знаний о заболевании, так и с фенотипическим разнообразием его форм. Ограниченные возможности ДНК-диагностики МВ в России и ее низкая доступность затрудняют и затягивают окончательную верификацию заболевания.
ЛИТЕРАТУРА
1. Муковисцидоз. Под ред. Н.И. Капранова, Н.Ю. Каширской. М.: ИД «МЕДПРАКТИКА-М», 2014, 672 с. ISBN 978-5-98803-314-1
2. Welsh M.J., Ramsey B.W., Accurso F.J., Cutting G.R. Cystic fibrosis. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill, 2001: 5121-88.
3. European cystic fibrosis society standards of care working group. Best practice guidelines. В редакции Alan R. Smith и Scott Bell, 2014.
4. Farell P.M., Rosenstein B.J., White T.B. et al. Cystic fibrosis foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report // J. Pediatr., 2008; 153 (2): S4-S14.
5. Красовский С.А., Каширская Н.Ю., Усачева М.В., Амелина Е.Л., Черняк А.В., Науменко Ж.К. Влияние возраста постановки диагноза и начала специфической терапии на основные клинико-лабораторные проявления заболевания у больных муковисцидозом // Вопросы современной педиатрии, 2014, т. 13, № 2, с. 36-43.
6. de Boeck K., Wilschanski M., Castellani C. et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax, 2006; 61: 627-635.
7. de Oronzo M.A. Hyperechogenic fetal bowel: an ultrasonographic marker for adverse fetal and neonatal outcome? // J. Prenat. Med., 2011 Jan-Mar; 5 (1): 9-13.
8. Bombieri C. et al. Recommendations for the classification of diseases as CFTR-related disorders // Journal of Cystic Fibrosis, 2011, vol. 10, suppl. 2; S86-S102.
9. Hall E., Lapworth R. Use of sweat conductivity measurements. Annals of Clinical Biochemistry, 2010; 47: 390-392.
10. Sands D., Oltarzewski M., Nowakowska A., Zybert K. Bilateral sweat tests with two different methods as a part of cystic fibrosis newborn screening (CF NBS) protocol and additional quality control. Folia Histochem Cystobiol., 2010 Sep 30; 48 (3): 358-65.
11. Sezer R.G., Aydemir G., Akcan A.B. et al. Nanoduct sweat conductivity measurements in 2664 patients: relationship to age, arterial blood gas, serum electrolyte profiles and clinical diagnosis // J. Clin. Med. Res., 2013 Feb; 5 (1): 34-41.
12. Петрова Н.В. Молекулярно-генетические и клинико-генотипические особенности муковисцидоза в российских популяциях. Автореф. дисс. докт. биол. наук. М., 2009, 42 с.
13. Derichs N., Sanz J., Von Kanel T. et al. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax, 2010 Jul; 65 (7): 594-9.
14. Servidoni M.F., Sousa M., Vinagre A.M. et al. Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterol., 2013 May 20; 13 (1): 91.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Здравствуйте , первый раз пишу тут , пишу от бессилия.
Неделю назад нам позвонил доктор и сказал , что пришли результаты скрининга Муковисцидоз результат сомнительный.
Вроде понимаю что это ошибка и просто был не правильный забор и все у нас будет хорошо , но все равно места себе не нахожу.
Делали мы не на тощак , никто ничего не говорил , как обычно покормила ребёнка и вот видимо из-за этого ложноположительный.
Кушает отлично , вес набирает тоже.
У кого был сомнительный результат И потом все было хорошо ?
Clinical complications in children with false-negative results in cystic fibrosis newborn screening
Katarzyna Zybert et al.
J Pediatr (Rio J).
2022 Jul-Aug.
Free PMC article
Abstract
Objective:
To present signs and symptoms and clinical course in cystic fibrosis patients with false-negative newborn screening (CF NBS).
Materials and methods:
All children presented in this paper were covered by CF NBS. The group of 1.869.246 newborns was screened in the Institute of Mother and Child in Warsaw within a period of 01.01.1999 — 31.05.2019. Screening protocols evolved over time from IRT/IRT to IRT/DNA/EGA.
Results:
The authors identified 11 patients with false-negative NBS, in whom CF was diagnosed based on clinical symptoms or the examination of siblings with positive CF NBS. In the study group, the diagnosis was made significantly later in comparison to positive CF NBS patients ranging from 2 months to 15 years of age. CF NBS strategy does not significantly affect the sensitivity of the screening.
Conclusion:
In the presence of clinical symptoms, additional diagnostics must be implemented, in spite of the negative screening results. At first, the sweat test should be conducted, followed by a DNA analysis of the most common mutations in the given population. The diagnostic process requires searching for CFTR mutations not typically associated with a high chloride concentration in sweat. Repetition of sweat chloride concentration enables the diagnosis in children whose initial chloride values in sweat are borderline, and no CF-causing mutations are detected. In strong clinical indications, the extension of DNA analysis (EGA) is recommended in order to identify rare CF variants. In children with meconium ileus, genetic analysis is mandatory.
Keywords:
Cystic fibrosis; Diagnosis; False-negatives; Newborn screening; Sweat test.
Copyright © 2021 Sociedade Brasileira de Pediatria. Published by Elsevier Editora Ltda. All rights reserved.
Conflict of interest statement
Conflicts of interest The authors declare no conflicts of interest.
Figures
Current newborn screening model in Poland (IRT/DNA/EGA).
Figure 1
Similar articles
-
Diagnosing cystic fibrosis in newborn screening in Poland — 15 years of experience.
Sands D, Zybert K, Mierzejewska E, Ołtarzewski M.
Sands D, et al.
Dev Period Med. 2015 Jan-Mar;19(1):16-24.
Dev Period Med. 2015.PMID: 26003066
-
Cystic fibrosis newborn screening: the importance of bloodspot sample quality.
Doull I, Course CW, Hanks RE, Southern KW, Forton JT, Thia LP, Moat SJ.
Doull I, et al.
Arch Dis Child. 2021 Mar;106(3):253-257. doi: 10.1136/archdischild-2020-318999. Epub 2020 Aug 28.
Arch Dis Child. 2021.PMID: 32859613
-
Diagnostic dilemmas resulting from the immunoreactive trypsinogen/DNA cystic fibrosis newborn screening algorithm.
Parad RB, Comeau AM.
Parad RB, et al.
J Pediatr. 2005 Sep;147(3 Suppl):S78-82. doi: 10.1016/j.jpeds.2005.08.017.
J Pediatr. 2005.PMID: 16202789
-
The diagnosis of cystic fibrosis.
De Boeck K, Vermeulen F, Dupont L.
De Boeck K, et al.
Presse Med. 2017 Jun;46(6 Pt 2):e97-e108. doi: 10.1016/j.lpm.2017.04.010. Epub 2017 May 31.
Presse Med. 2017.PMID: 28576637
Review.
-
Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report.
Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW 3rd; Cystic Fibrosis Foundation.
Farrell PM, et al.
J Pediatr. 2008 Aug;153(2):S4-S14. doi: 10.1016/j.jpeds.2008.05.005.
J Pediatr. 2008.PMID: 18639722
Free PMC article.
References
-
-
Crossley JR, Elliott RB, Smith PA. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet. 1979;1:472–474.
—
PubMed
-
-
-
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073.
—
PubMed
-
-
-
Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065.
—
PubMed
-
-
-
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080.
—
PubMed
-
-
-
Barben J, Castellani C, Dankert-Roelse J, Gartner S, Kashirskaya N, Linnane B, et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J Cyst Fibros. 2017;16:207–213.
—
PubMed
-
MeSH terms
Substances
LinkOut — more resources
-
Full Text Sources
- Elsevier Science
- Europe PubMed Central
- PubMed Central
- Sociedade Brasileira de Pediatria
-
Medical
- Genetic Alliance
- MedlinePlus Health Information
-
Research Materials
- NCI CPTC Antibody Characterization Program
-
Miscellaneous
- NCI CPTAC Assay Portal
Обновлено: 23.06.2023
Скрининг на муковисцидоз. Пренатальная диагностика
В настоящее время невозможно объяснить высокую частоту мутантных аллелей CFTR, наблюдаемую в европеоидных популяциях и составляющую 1 на 50. Болезнь значительно реже встречается в других популяциях, хотя описана у американских индейцев, афроамериканцев и монголоидов (например, среди жителей Гавайских островов азиатского происхождения с частотой приблизительно 1 на 90 000).
Аллель F508 — единственный общий фактически для всех европеоидных популяций из обнаруженных до настоящего времени. Анализ гаплотипов европеоидных популяций указывает, что аллель F508, вероятно, имеет единственное начало. Частота этого аллеля среди всех мутантных аллелей значительно изменяется в разных европейских популяциях, с 88% в Дании до 45% в южной Италии.
В популяциях, в которых частота аллеля F508 составляет приблизительно 70%, около 50% пациентов — гомозиготны по аллелю F508; еще 40% — генетические компаунды F508 и другого мутантного аллеля. Кроме того, приблизительно 70% носителей муковисцидоза имеют мутацию F508.
За исключением F508, остальные мутации в локусе CFTR встречаются редко, хотя в специфических популяциях некоторые аллели могут встречаться чаще других.
Популяционный скрининг на муковисцидоз
В настоящее время муковисцидоз соответствует большинству критериев для программы скрининга новорожденных, за исключением того, что пока не ясно, насколько ранняя идентификация больных новорожденных улучшает долгосрочный прогноз.
Тем не менее преимущества ранней диагностики (например, улучшенное питание с обеспечением ферментами поджелудочной железы) позволили осуществлять программы неонатального скрининга. Хотя обычно считают, что скрининг носительства нецелесообразен до тех пор, пока не будут обнаруживаться по крайней мере 90% мутаций (в настоящее время — почти 85%), в Соединенных Штатах в течение нескольких лет осуществляют скрининг среди семейных пар на уровне частных медицинских методов.
Генетический анализ семей пациентов и пренатальная диагностика муковисцидоза
Высокая частота аллеля F508 информативна, когда ДНК диагностику проводят пациенту без семейной истории муковисцидоза. Для того чтобы определить статус членов семьи для подтверждения болезни (например, у новорожденного или сибса с неоднозначной симптоматикой), диагностировать носительство или провести пренатальную диагностику можно, используя идентификацию аллеля AF508 в комбинации с панелью из 22 менее частых, но и не редких мутаций, предлагаемую Американским колледжем медицинской генетики.
При условии хорошего знания мутаций муковисцидоза в популяции прямое обнаружение мутации — метод выбора для генетического анализа. Если при отсутствии известной специфической мутации использовать анализ сцепления, точная диагностика возможна фактически во всех семьях.
При беременности с риском 1 к 4 метод выбора — пренатальная ДНК-диагностика на сроке от 10 до 12 нед в тканях, получаемых при БВХ. Биохимические методы пренатальной диагностики, основанные на измерении пищеварительных ферментов (например, щелочной фосфатазы) в амниотической жидкости, имеют высокие ложноположительные показатели, и их больше не используют.
Молекулярная генетика и лечение муковисцидоза. В настоящее время лечение муковисцидоза направлено на борьбу с легочной инфекцией и улучшение питания. Повышение знаний о молекулярном патогенезе может сделать возможным разработку фармакологических вмешательств с целью непосредственно скорректировать аномальный биохимический фенотип.
Кроме того, при муковисцидозе может оказаться возможной генотерапия, но пока есть множество трудностей.
Видео этиология, патогенез муковисцидоза
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Муковисцидоз (Расширенный поиск частых мутаций в гене CFTR (30 шт.))
Лабораторная диагностика муковисцидоза безопасно оценивает риск наличия данного генетического заболевания у пациента, исследование проводится в лаборатории «Проген», необходима венозная кровь пациента, анализ крови позволит проанализировать гены (CFTR) отвечающие за данное заболевание и выдать заключение.
- Навигация
- Муковисцидоз, что это?
- Как проявляется болезнь муковисцидоз?
- Какие симптомы у муковисцидоза?
- Есть ли лекарство от муковисцидоза?
Муковисцидоз, что это?
Муковисцидоз представляет собой серьезное генетическое заболевание, которое в основном поражает дыхательную и пищеварительную системы, но также затрагивает и другие внутренние органы.
Это происходит из — за мутировавшего, измененного гена, который называется CFTR (муковисцидоз трансмембранного регулятора), мутировавший ген вызывает производство слишком густой слизи в дыхательных путях и препятствует выводу из организма воды, соли из клеток, а также препятствует выводу бактерий и частиц. Эта слизь закрывает бронхи и приводит к повторным респираторным инфекциям, закупоривает поджелудочную железу и препятствует попаданию ферментов поджелудочной железы в кишечник, в результате чего пища не может нормально перевариваться и усваиваться.
Хотя степень поражения также сильно различается от человека к человеку, стойкая легочная инфекция и воспаление, которые вызывают прогрессирующее разрушение легочной ткани, являются основной причиной заболеваемости у пациентов с муковисцидозом.
Как проявляется болезнь муковисцидоз?
При наличии данного заболевания появляются:
- Трудности с перевариванием жиров, белков, крахмала;
- Дефицит жирорастворимых витаминов;
- Прогрессирующая потеря функции легких.
Заболевание никоим образом не вредит интеллектуальным способностям и не проявляется в физическом аспекте, ни при рождении, ни в более позднем возрасте, поэтому его называют «невидимой болезнью».
Какие симптомы у муковисцидоза?
Нарушение функции или отсутствие белка CFTR влияет на все секретирующие железы слизистой оболочки, что приводит к недостатку хлора и воды в секретах. Слизистые выделения, очень вязкие, имеют тенденцию застаиваться, вызывая закупорку пораженных органов, в основном бронхов, кишечника и поджелудочной железы.
В бронхиальном дереве (степень поражения которого является основной причиной тяжести заболевания) запускается порочный круг воспаления-инфекции, что в конечном итоге приводит к разрушению легочной ткани.
Муковисцидоз — это заболевание, поражающее многие органы и вызывающее множество симптомов, в том числе:
- Легкие;
- Органы пищеварения;
- Уши, горло, нос и носовые пазухи;
- Кости;
- Печень.
Эти поражения органов приводят к следующей симптоматике:
- Появляется кашель с мокротой или слизью, частые респираторные инфекции и нарастающая одышка;
- Гайморит;
- Проблемы усваивания пищи, ведущие к недостатку питания;
- Диабет;
- Цирроз или рубцевание печени;
- Снижение детородной функции.
Есть ли лекарство от муковисцидоза?
Лечение этого заболевания в последние годы продвинулось далеко вперед.
Основные методы лечения муковисцидоза направлены на облегчение симптомов и улучшение качества жизни пациентов.
Недавно в США был представлен препарат применяющийся для лечения муковисцидоза.
Препарат отпускаемый по рецепту врача, который используется для лечения муковисцидоза (CF) у пациентов в возрасте от 2 лет и старше, у которых есть две копии мутации F508del (F508del / F508del) в их гене CFTR. Не следует применять данный препарат людям, кроме тех, у которых есть две копии мутации F508del в гене CFTR.
Важно! Прежде чем принимать препараты, необходимо провести генетическое исследование, лабораторную диагностику муковисцидоза, подтвердить наличие мутации в гене CFTR и получить официальный медицинский диагноз у врача. В лаборатории «Проген» можно выявить факт носительства мутации и определить риски.
Доступные тесты
Безопасно оценивает риск основных хромосомных заболеваний плода (3 синдрома + определение пола плода) с 10 недели беременности.
Занятие 2. Диагностика муковисцидоза. Диспансерное наблюдение.
Анализ крови, включая определение иммунного реактивного трипсина через несколько дней после рождения, является одним из методов диагностики определенных заболеваний, включая муковисцидоз. Однако, этот первый анализ только позволяет заподозрить заболевание. Оно должно быть подтверждено дополнительными исследованиями. Чаще всего диагноз ставится по анализам пота. Он состоит в определении количества соли в поте. В случае муковисцидоза эта концентрация в 2-5 раз превышает норму. Анализ можно считать надежным, если он проводится в очень строгих условиях в специализированном центре.
Если же результат не является убедительным, то врач может провести новое исследование. Этот анализ очень быстрый, безболезненный и результаты становятся известны в течение 24 часов. После этих исследований проводят генетический анализ крови. Данный анализ позволяет определить наличие мутации.
Неонатальный скрининг
Диагностике муковисцидоза сегодня способствует скрининг новорожденных, выполняемый в РФ с 2006г. Слово «скрининг» (с английского языка- «прочёсывать») означает обследование всех здоровых людей, для выявления некоторых скрытых и значимых заболеваний.
Целью неонатального скрининга является выявление некоторых заболеваний, которые, при раннем их обнаружении, можно лечить с отличными результатами. Это болезни, которые уже присутствуют с рождения, но при этом могут не проявляться в первые месяцы жизни, это такие заболевания, как фенилкетонурия, врождённый гипотиреоз и муковисцидоз. Если при фенилкетонурии (1 случай на 20 000 новорожденных) и врожденного гипотиреоза (1 на 2500) раннее обнаружение позволяет начать раннее лечение и приводит к выздоровлению, то при муковисцидозе проведение скрининга позволяет начать раннее и своевременное лечение, которое хотя и не приводит к выздоровлению, однако, может продлить жизнь и улучшить ее качество.
Неонатальный скрининг основан на определении в крови уровня определённого белка, фермента поджелудочной железы- трипсина (анализ на иммунореактивный трипсин, или ИРТ). Уровень трипсина определяется в капельке крови, взятой из пятки, на промокательную бумагу, на 3 й -4 й день поле рождения. Если уровень ИРТ превышает определённый нормальный порог (это определённый показатель у каждой скрининговой лаборатории), то высказывается подозрение на наличие заболевания. В этом случае проводится повторный анализ на 28 день жизни: если результат нормальный, то вопрос закрывается, если же повторно положительный, то проводится потовый тест и затем, возможно, генетическое исследование. Если генетический анализ идентифицирует наличие двух мутаций гена CFTR, то диагноз муковисцидоза является бесспорным.
Обычно, для постановки диагноза решающее значение имеет потовый тест, даже если до этого было выявлено наличие двух мутаций гена CFTR. Если потовый тест указывает на наличие муковисцидоза, обычно организуется короткая госпитализация ребёнка для определения особенностей болезни (степень вовлечения в процесс лёгких, наличие или отсутствие недостаточности поджелудочной железы и так далее) и для назначения программы лечения
♦ Что означает положительный результат скрининга?
Положительный результат скрининга говорит о том, что есть подозрение на муковисцидоз. Это подозрение может быть подтверждено («истинный положительный») или опровергнуто («ложный положительный») путем последующих исследований. На самом деле, неонатальный скрининг на муковисцидоз даёт некоторое количество положительных результатов у абсолютно здоровых детей: из каждых 3-6 случаев положительного результата только один оказывается действительно больным муковисцидозом, а оставшиеся — это ложноположительные результаты. Это происходит потому, что повышенный уровень трипсина в крови не является узкоспецифическим маркером для муковисцидоза и он может быть повышен не только при муковисцидозе, или будучи повышенным при рождении, если ребёнок здоров, он нормализуется в течение последующих дней или недель. Напротив, у больного муковисцидозом он остаётся повышенным длительно, месяцами. Поэтому необходимы углубленные обследования, о которых было сказано выше. Одно из них – это генетическое исследование. Оно может определить наличие мутаций гена CFTR, не определяя их количество — одна или две. Но при наличии только одной мутации, ребенок может быть всего лишь здоровым носителем, при наличии же двух мутаций гена CFTR, несомненно, ребёнок будет болен, и потовый тест необходим для подтверждения окончательного диагноза. Поэтому так важно выполнить потовый тест как можно быстрее, чтобы внести определённость, успокоить родителей и при необходимости начать соответствующее лечение.
♦ Что означает отрицательный результат скрининга? Отрицательный результат скрининга говорит о том, что трипсиновый тест не вызвал подозрения на наличие муковисцидоза. Почти всегда это говорит об отсутствии муковисцидоза («истинный отрицательный» результат). Дети с «истинным отрицательным результатом» представляют подавляющее большинство случаев. Однако изредка при скрининге бывают «ложноотрицательные» результаты. Точно сказать нельзя, но в 3-5% больные дети не распознаются при скрининге. Это больные муковисцидозом дети, у которых трипсиновый тест не показал наличие заболевания, но диагноз устанавливается в последующие годы жизни, на основании проявлений, которые позволяют заподозрить наличие болезни. Таким образом, если результат скрининга отрицательный, весьма маловероятно, что ребенок болен муковисцидозом, но теоретически небольшая вероятность этого всё же остаётся, и при наличии подозрительных признаков всегда лучше сделать потовый тест.
Этапы неонатального скрининга в Российской Федерации
На 3-4 день у доношенного (на 7-8–й – у недоношенного) новорожденного – определение ИРТ
в высушенной капле крови
Диагностика муковисцидоза
В последнее время в мире стали проводить генетические анализы для определения муковисцидоза у младенцев. У ребёнка берут анализ крови, если выясняется, что уровень иммунореактивного трипсина находится на высоких отметках, новорожденный находится в группе риска. Но, для подтверждения болезни или её отсутствия дополнительно проводится исследование солей в потовой жидкости. Некоторые родители спрашивают: для чего нужно при подтверждённом заболевании проходить исследование? Чтобы ответить на этот вопрос, необходимо более детально рассмотреть эту болезнь и узнать к чему она может привести.
Муковисцидоз — что это за болезнь?
Муковисцидоз – это тяжёлое генетическое заболевание, вызванное мутацией гена CFTR в седьмой хромосоме, которое не поддаётся лечению. Мутация приводит к тому, что белок регулирующий передвижение электролитов через структуру клетки вырабатывается в небольших количествах или перестаёт вырабатываться совсем.
В результате нарушения функции синтезируемого белка, слизь, вырабатываемая дыхательными органами, а также желудочно-кишечным трактом, становится вязкой, густой, что приводит к поражению этих органов и в них развиваются склеротические изменения. Заразиться муковисцидозом невозможно, так как с этим заболеванием уже рождаются, и болезнь поражает с одинаковой частотой, как девочек, так и мальчиков.
Симптомы и причины муковисцидоза у детей
Больные начинают часто болеть бронхитом, воспалением лёгких, также может развиться цирроз печени или желчнокаменная болезнь вызванная застоем желчи.
К частым проявлениям (симптомам заболевания) относят:
- хронический влажный кашель;
- прослушиваемые хрипы в легких;
- часто повторяющиеся заболевания по типу ОРВИ, пневмания и бронхит;
- слабость и вялость.
Люди, имеющие только один дефектный ген, называются носителями. У ребёнка при обследовании могут выявить это заболевание, только если родители являются носителями мутаций.
Неинвазивный пренатальный тест ДНК и ранее определение муковисцидоза
У носителей дефектного гена риск рождения ребёнка с диагнозом муковисцидоз возрастает на 25% с каждой последующей беременностью. Чтобы этого не произошло, нужно выявить, какой именно дефект гена CFTR спровоцировал заболевание у ребёнка.
Самый простой и дешёвый способ – это пройти исследование на различные виды мутации, которых существует около тридцати. Если по результатам теста выявится один дефект, нужно будет исследовать всю последовательность гена методом секвенирования. Этот тест позволяет выявить все существующие мутации.
Это необходимо сделать для того, чтобы знать, носителями какого вида мутаций являются родители и при необходимости во время беременности или экстракорпоральном оплодотворении исследовать плод на их наличие. Есть несколько методов диагностики моногенных заболеваний, в том числе муковисцидоза:
Перед пересадкой эмбриона в полость матки часть его клеток исследуют на предмет носительства генетических заболеваний. После диагностики выдается заключение о пригодности переноса обследованного эмбриона.
— Во время беременности. В случае серьезных отклонений у плода по хромосомным аномалиям, на десятой-двенадцатой неделе беременности производится забор плаценты посредством прокола передней брюшной полости.
Далее материал проверяют на носительство генных мутаций, если они не обнаружены или выявлена одна мутация, можно не опасаться, что ребёнок родится больным. В ситуации, когда выявлено два дефектных гена, диагноз считается подтверждённым, и родители стоят перед выбором рождения ребёнка с муковисцидозом или прерывания беременности.
— Во время беременности, безопасный вариант. Также существует безопасный и абсолютно нетравматичный метод исследования генетического здоровья будущего ребенка во время беременности — неинвазивный пренатальный тест.
В России только один европейский тест исследует ДНК плода на 100 моногенных заболеваний, это тест VERAgene, производство Кипр.
Тест проводится уже с 10 недель беременности и для проведения анализа требуется только венозная кровь матери. Точность исследования такая же как и при инвазивных процедурах.
Искренне надеемся, что со временем появятся новые лекарства способные регулировать работу генов, и назначение медикамента будет зависеть от того, какой тип мутации гена у ребёнка.
Чтобы пройти тест VERAgene обратитесь в лабораторию Медикал Геномикс. Также в случае положительного результата будет возможность проконсультироваться с практикующим врачом-генетиком нашего медицинского центра бесплатно.
Скрининг на муковисцидоз. Пренатальная диагностика
Комплексное генетическое исследование, которое позволяет выявить 28 наиболее часто встречающихся на территории России мутаций гена CFTR, приводящих к развитию тяжелого наследственного заболевания муковисцидоза.
Метод исследования
Какой биоматериал можно использовать для исследования?
Венозную кровь, буккальный (щечный) эпителий.
Как правильно подготовиться к исследованию?
Специальной подготовки не требуется.
Общая информация об исследовании
Муковисцидоз (синоним – кистозный фиброз) – одно из наиболее распространенных аутосомно-рецессивных наследственных заболеваний человека. Он характеризуется нарушением функции эпителия дыхательных путей, кишечника, поджелудочной железы, потовых и половых желез.
Причиной развития муковисцидоза являются мутации в гене CFTR (cystic fibrosis transmembrane regulator), кодирующем АТФ-связывающий белок, который формирует канал для ионов хлора в клеточных стенках. Мутации приводят к нарушению транспорта электролитов и ионов хлора через мембраны эпителиальных клеток, что сопровождается усилением секреции густой слизи и закупоркой выводящих протоков различных желез.
Существует несколько форм муковисцидоза:
- смешанная (поражаются одновременно органы дыхания и пищеварительный тракт);
- бронхолегочная (поражаются преимущественно органы дыхания);
- кишечная (поражается преимущественно желудочно-кишечный тракт);
- мекониевая непроходимость кишечника;
- атипичные формы, связанные с изолированными поражениями отдельных желез внешней секреции.
В настоящее время в РФ диагноз «муковисцидоз» ставится одному из 9 000 новорождённых (для сравнения: в Европе муковисцидоз диагностируется с частотой 1 : 2 000 — 3 000 новорождённых). Однако принятая в России форма массового скрининга новорождённых несовершенна и иногда не позволяет выявить заболевание на доклинической стадии.
В каждой клетке нашего организма имеется две копии гена CFTR. Одна копия достается от отца, а вторая от матери. Заболевание муковисцидоз аутосомно-рецессивное, т. е. развивается оно только при условии, что ребенок получает и от отца, и от матери мутантный ген CFTR. При этом родители, у которых вторые копии гена CFTR нормальные, не страдают муковисцидозом и порой даже не догадываются о его носительстве. По статистике, в европейской популяции носителем мутаций гена CFTR в среднем является каждый 25-й человек.
Насчитывается примерно одна тысяча различных мутаций в гене CFTR. Они встречаются с различной частотой в разных популяциях. Некоторые нарушения в гене могут не иметь никаких проявлений. Но большая часть мутаций вызывает патологический эффект, т. к. приводит к нарушению функционирования белка.
В данное комплексное исследование включен анализ гена CFTR на 28 мутаций, наиболее распространенных на территории РФ, Восточной Европы и Скандинавии и связанных с развитием тяжелых клинических форм муковисцидоза. Исследование позволяет выявлять до 95 % всех возможных больных, что существенно превышает разрешающие способности утвержденного в России скрининга.
Исследование поможет не только подтвердить или опровергнуть диагноз «муковисцидоз», но и выявить носительство мутации у здоровых людей. Особенно важно проводить генетическое тестирование в семьях, в которых есть больные муковисцидозом, поскольку у пары, где оба родителя являются носителями мутаций, вероятность рождения больного ребенка составляет 25 %.
До сих пор муковисцидоз считается неизлечимым заболеванием, но ранняя диагностика и адекватная терапия значительно улучшают прогноз заболевания и продляют пациенту жизнь.
Читайте также:
- Акцелерации. Спородические, периодические акцелерации. Децелерации. Спородические, периодические децелерации.
- Лимфоцитарный лейкоцитоз
- Синтез фосфолипидов в клетке — цикл Кеннеди
- Лучевые признаки геморрагической кисты яичника
- Деформации и дистрофии ногтей
СЛУЧАЙ ЛОЖНООТРИЦАТЕЛЬНЫХ ПОТОВЫХ ПРОБ У РЕБЕНКА С МУКОВИСЦИДОЗОМ В АЛТАЙСКОМ КРАЕ
Алтайский государственный медицинский университет, г. Барнаул Близнюк Е.А., Колесникова О.И., Сероклинов В.Н.
A CASE OF FALSE-NEGATIVE SWEAT SAMPLES IN A CHILD WITH CYSTIC FIBROSIS IN ALTAI KRAI
Altai State Medical University, Barnaul Bliznyuk E.A., Kolesnikova O.I., Seroklinov V.N.
Актуальность
Муковисцидоз (МВ), или кистозный фиброз — частое моногенное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза, характеризующееся поражением экзокринных желез систем органов дыхания и пищеварения, наследуемое по аутосомно-рецессивному типу. Своевременная диагностика МВ, обеспечивающая в большинстве случаев раннее начало терапии, в том числе на доклиническом этапе, улучшает прогноз заболевания, повышает эффективность лечения, позволяет предупредить развитие тяжелых осложнений, значительного отставания в физическом развитии, а в ряде случаев и необратимых изменений в легких. С 2006 года в Алтайском крае проводится скрининг новорожденных на МВ. Это позволяет поставить диагноз «муковисцидоз» в первые 3-4 месяца жизни ребенка. Надежным методом диагностики МВ является потовая проба.
Цель исследования: Изучить случаи ложноотрицательных потовых проб у детей с муковисцидозом, родившихся в 2019-2020 гг. в Алтайском крае.
Пациенты и методы: Определение иммунореактивного трипсина в сухих пятнах крови новорожденных. Определение проводимости пота на аппарате «Нанодакт» у детей с неонатальной гипертрипсиногенемией. Молекулярно-генетическое исследование 50-ти частых мутаций гена CFTR (трансмембранного регуляторного белка муковисцидоза) у детей с МВ.
Результаты
С января 2019 г. по декабрь 2020 г. в Алтайском крае обследовано 40456 новорожденных по скринингу на МВ. Неонатальная гипертрипсиногенемия (положительный тест на 4-5 день жизни и положительный ретест на 21-28 день жизни на иммунореактивный трипсин в сухом пятне крови) выявлена у 17 новорожденных. Среди этих 17 детей с неонатальной гипертрипсиногенемией
муковисцидоз выявлен у 6. Средний возраст диагностики муковисцидоза у 5 детей составил 3,5 месяца. У одной девочки с неонатальной гипертрипсиногениемией, родившейся в 2019 году, диагноз «муковисцидоз» установлен в возрасте 1 год 1 месяц. Описание этого случая поздней диагностики муковисцидоза приведено ниже.
Девочка, 1 год 1 месяц, поступила в эндокринологическое отделение (пульмонологический профиль) КГБУЗ «Алтайский краевой клинический центр охраны материнства и детства» (АККЦОМД) с жалобами на продуктивный приступообразный кашель, усиливающийся в ночное время, дыхание с дистантными хрипами, плохую прибавку веса с рождения.
Из анамнеза заболевания: Результат неонатального скрининга на МВ положительный (анализ крови на иммунореактивный трипсин: первичный тест -137 нг/мл — 129 нг/мл (норма до 65 нг/мл), ретест — 104,5 нг/мл (норма до 40 нг/мл). В связи с этим в 1 -месячном возрасте проведен потовый тест в биохимической лаборатории медико-генетической консультации Диагностического центра Алтайского края, который оказался отрицательным (электропроводимость пота -47 ммоль/л NaCl). С 6-месячного возраста отмечались приступообразный кашель, периодические дистантные хрипы. Больная 2 раза лечилась в стационаре по месту жительства с диагнозом «Острый бронхит, затяжное течение». На фоне проводимой терапии динамика слабоположительная, сохранялся кашель и эпизоды шумного дыхания. Ребенок обследован в КГБУЗ «АККЦОМД» в 11 месяцев. Убедительных данных за МВ не установлено (из трех проведенных потовых проб положительная одна: электропроводимость пота 31 — 127 — 59 ммоль/л NaCl). При повторной госпитализации в возрасте 1 год 1 месяц из трех проведенных потовых проб положительных две (электропроводимость пота 45 — 86 — 81 ммоль/л NaCl). Установлен диагноз: Основной: Кистозный фиброз с другими проявлениями (Е84.8). Муковисцидоз, смешанная форма, среднетяжелое течение. Обструктивный бронхит средней степени тяжести. Первичный высев Pseudomonas aeruginosa, Staph. aureus. Генетический диагноз не установлен. Осложнения: Хроническая панкреатическая недостаточность. Легкая белково-энергетическая недостаточность.
В дальнейшем диагноз подтвержден с помощью ДНК-диагностики в научно-исследовательском институте медицинской генетики (г. Томск): молекулярно-генетическое исследование 50-ти частых мутаций гена CFTR от 15.12.2020 г. -генотип F508del/CFTRdele2,3.
Вывод
Запоздалая диагностика МВ у ребенка с неонатальной гипертрипсиногенемией была обусловлена неоднократными
ложноотрицательными потовыми пробами при наличии клинической картины заболевания. Отрицательные значения потовых проб нельзя объяснить наличием в генотипе мягких мутаций гена CFTR, при которых частично сохраняется функция хлорного канала. Ведь у девочки выявлены две тяжелые мутации гена CFTR, серьезно нарушающие работу хлорного канала. Возможно, отрицательные потовые пробы связаны с наличием в геноме девочки генов-модификаторов, влияющих на работу хлорных каналов в потовых железах ребенка. В случаях затруднения диагностики МВ (сомнительные потовые пробы, чередование отрицательных и положительных потовых проб) можно рекомендовать более широкое использование молекулярно-генетических методов исследования (ДНК-диагностика на наиболее частые мутации гена муковисцидозного трансмембранного регулятора, секвенирование гена МВ).
Список литературы:
1. Кондратьева Е.И., Шерман В.Д., Капранов Н.И., Каширская Н.Ю. Актуальные вопросы диагностики муковисцидоза. ПРАКТИКА ПЕДИАТРА. 2015;2:20-27.
2. Клинические рекомендации. Кистозный фиброз (муковисцидоз). МЗ РФ. 2020;108 с.
3. Национальный консенсус (2-е издание) «Муковисцидоз: определение, диагностические критерии, терапия» — 2019. Под редакцией Е.И. Кондратьевой, Н.Ю. Каширской, Н.И. Капранова. М.: ООО «Компания БОРГЕС». 2018;356 с.